Kurzbeschreibung:
Die Sichelzellanämie gehört zu den Hämoglobinopathien. Ein Gendefekt führt zur Bildung einer abnormen b-Kette, dem so genannten Hämoglobin S. Desoxigeniertes HbS neigt dazu, in langen Polymeren zu flüssigen Kristallen zu aggregieren. Die Erythrozyten verlieren dadurch ihre Deformierbarkeit und nehmen die Sichel-Form an. Diese Erythrozyten werden im retikulo-endothelialen System sequestriert (extravasale Hämolyse) oder führen in kleinen Gefässen zu Verschlüssen (Infarkte). Die Sichelzellanämie kommt vor allem bei Menschen aus Schwarzafrika und der Karibik vor. Sie bietet einen gewissen Schutz gegen Malaria. Die Tatsache, dass Sauerstoffabschluss zur Sichelung der Erythrozyten führt, wird im Labor für den Sichelzelltest genutzt. Die definitive Diagnose wird mittels Hämoglobin-Elektrophorese gestellt.
Klinisches Bild:
Bei der Sichelzellanämie handelt es sich um die homozygote Form. Die heterozygote From wird Sichelzell-Trait genannt. Trotz einer Hämoglobinkonzentration zwischen 60 und 100 g/L sind die Anämie-Symptome gering, da beim HbS der Sauerstoff früh dissoziiert. Charakteristisch für die Sichelzellanämie sind die Sichelzellkrisen. Hierbei handelt es sich um akute, schmerzhafte Episoden, die durch vaskuläre Verschlüsse verursacht werden. Meist werden sie durch Infektionen oder Kälteexposition ausgelöst. Von diesen Gefässverschlüssen können alle Organe betroffen sein. Typisch folgen daraus unterschiedliches Längenwachstum von Fingern und Zehen. Cholelithiasis als Folge der chronischen Hämolyse tritt häufig auf. Wie bei der Sphärozytose können Infektionen durch Parvovirus B19 zu aplastischen Krisen führen.
Hämatologie:
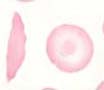 Im Blutausstrich finden sich die charakteristischen Sichelzellen sowie Targetzellen. Bei Erwachsenen liegt oft auf Grund der rezidivierenden Infarkte eine funktionelle Asplenie vor. Entsprechend können Howell-Jolly-Körper und Pappenheimer Körper auftreten.
Im Blutausstrich finden sich die charakteristischen Sichelzellen sowie Targetzellen. Bei Erwachsenen liegt oft auf Grund der rezidivierenden Infarkte eine funktionelle Asplenie vor. Entsprechend können Howell-Jolly-Körper und Pappenheimer Körper auftreten.
Inhaltsverzeichnis